HCA Project
The Human Cell Atlas (HCA) is a large project that aims to learn from and map every cell type in the human body. The project extracts spatial and molecular characteristics in order to understand cellular function and networks. It is an international collaborative that charts healthy cells in the human body at all ages. There are about 37.2 trillion cells in the human body. To read more about the project, head over to their website at https://www.humancellatlas.org.
CELLxGENE
CELLxGENE is a database and a suite of tools that help scientists to find, download, explore, analyze, annotate, and publish single cell data. It includes several analytic and visualization tools to help you to discover single cell data patterns. To see the list of tools, browse to https://cellxgene.cziscience.com/.
CELLxGENE | Census
The Census provides efficient computational tooling to access, query, and analyze all single-cell RNA data from CZ CELLxGENE Discover. Using a new access paradigm of cell-based slicing and querying, you can interact with the data through TileDB-SOMA, or get slices in AnnData or Seurat objects, thus accelerating your research by significantly minimizing data harmonization at https://chanzuckerberg.github.io/cellxgene-census/.
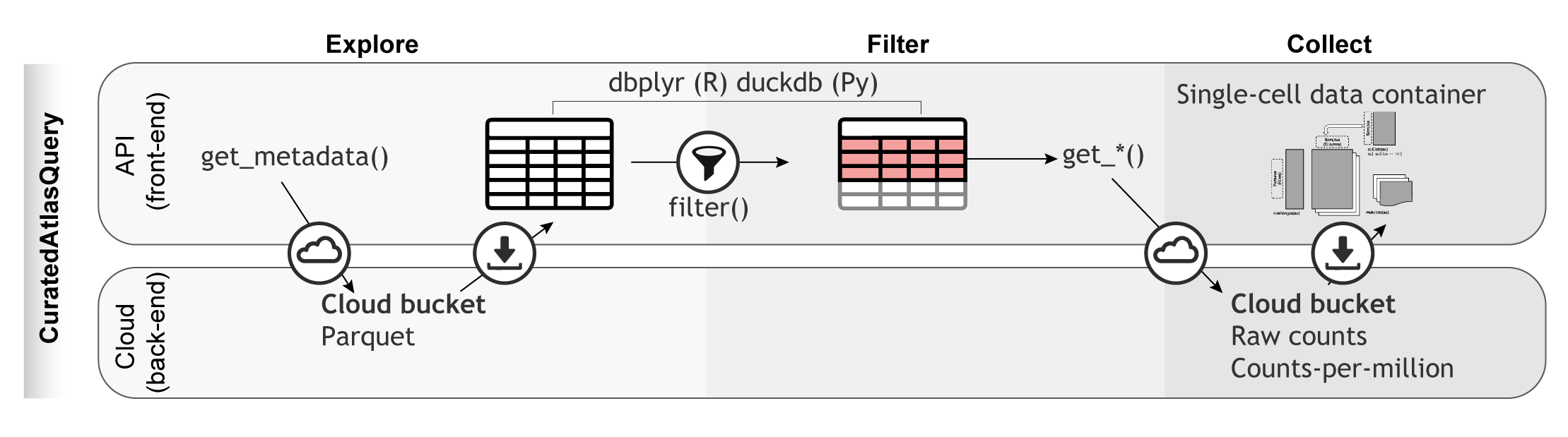
The CuratedAtlasQueryR Project
To systematically characterize the immune system across tissues, demographics and multiple studies, single cell transcriptomics data was harmonized from the CELLxGENE database. Data from 28,975,366 cells that cover 156 tissues (excluding cell cultures), 12,981 samples, and 324 studies were collected. The metadata was standardized, including sample identifiers, tissue labels (based on anatomy) and age. Also, the gene-transcript abundance of all samples was harmonized by putting values on the positive natural scale (i.e. non-logarithmic).
To model the immune system across studies, we adopted a consistent immune cell-type ontology appropriate for lymphoid and non-lymphoid tissues. We applied a consensus cell labeling strategy between the Seurat blueprint and Monaco (2019) to minimize biases in immune cell classification from study-specific standards.
CuratedAtlasQueryR supports data access and programmatic
exploration of the harmonized atlas. Cells of interest can be selected
based on ontology, tissue of origin, demographics, and disease. For
example, the user can select CD4 T helper cells across healthy and
diseased lymphoid tissue. The data for the selected cells can be
downloaded locally into popular single-cell data containers. Pseudo bulk
counts are also available to facilitate large-scale, summary analyses of
transcriptional profiles. This platform offers a standardized workflow
for accessing atlas-level datasets programmatically and
reproducibly.
Data Sources in R / Bioconductor
There are a few options to access single cell data with R / Bioconductor.
| Package | Target | Description |
|---|---|---|
| hca | HCA Data Portal API | Project, Sample, and File level HCA data |
| cellxgenedp | CellxGene | Human and mouse SC data including HCA |
| CuratedAtlasQueryR | CellxGene | fine-grained query capable CELLxGENE data including HCA |
Installation
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("stemangiola/CuratedAtlasQueryR")HCA Metadata
The metadata allows the user to get a lay of the land of what is available via the package. In this example, we are using the sample database URL which allows us to get a small and quick subset of the available metadata.
metadata <- get_metadata(remote_url = CuratedAtlasQueryR::SAMPLE_DATABASE_URL)Get a view of the first 10 columns in the metadata with
glimpse
## Rows: ??
## Columns: 10
## Database: DuckDB 0.8.1 [unknown@Linux 5.15.0-1041-azure:R 4.3.0/:memory:]
## $ cell_ <chr> "TTATGCTAGGGTGTTG_12", "GCTTGAACATGG…
## $ sample_ <chr> "039c558ca1c43dc74c563b58fe0d6289", …
## $ cell_type <chr> "mature NK T cell", "mature NK T cel…
## $ cell_type_harmonised <chr> "immune_unclassified", "cd8 tem", "i…
## $ confidence_class <dbl> 5, 3, 5, 5, 5, 5, 5, 5, 5, 5, 5, 1, …
## $ cell_annotation_azimuth_l2 <chr> "gdt", "cd8 tem", "cd8 tem", "cd8 te…
## $ cell_annotation_blueprint_singler <chr> "cd4 tem", "cd8 tem", "cd8 tcm", "cl…
## $ cell_annotation_monaco_singler <chr> "natural killer", "effector memory c…
## $ sample_id_db <chr> "0c1d320a7d0cbbc281a535912722d272", …
## $ `_sample_name` <chr> "BPH340PrSF_Via___transition zone of…A note on the piping operator
The vignette materials provided by CuratedAtlasQueryR
show the use of the ‘native’ R pipe (implemented after R version
4.1.0). For those not familiar with the pipe operator
(|>), it allows you to chain functions by passing the
left-hand side (LHS) to the first input (typically) on the right-hand
side (RHS).
In this example, we are extracting the iris data set
from the datasets package and ‘then’ taking a subset where
the sepal lengths are greater than 5 and ‘then’ summarizing the data for
each level in the Species variable with a
mean. The pipe operator can be read as ‘then’.
data("iris", package = "datasets")
iris |>
subset(Sepal.Length > 5) |>
aggregate(. ~ Species, data = _, mean)## Species Sepal.Length Sepal.Width Petal.Length Petal.Width
## 1 setosa 5.313636 3.713636 1.509091 0.2772727
## 2 versicolor 5.997872 2.804255 4.317021 1.3468085
## 3 virginica 6.622449 2.983673 5.573469 2.0326531Summarizing the metadata
For each distinct tissue and dataset combination, count the number of datasets by tissue type.
## # Source: SQL [?? x 2]
## # Database: DuckDB 0.8.1 [unknown@Linux 5.15.0-1041-azure:R 4.3.0/:memory:]
## tissue n
## <chr> <dbl>
## 1 transition zone of prostate 2
## 2 peripheral zone of prostate 2
## 3 heart left ventricle 7
## 4 lung 4
## 5 blood 17
## 6 bone marrow 4
## 7 kidney 8
## 8 renal medulla 6
## 9 cortex of kidney 7
## 10 renal pelvis 4
## # ℹ more rowsColumns available in the metadata
## [1] "cell_" "sample_"
## [3] "cell_type" "cell_type_harmonised"
## [5] "confidence_class" "cell_annotation_azimuth_l2"
## [7] "cell_annotation_blueprint_singler" "cell_annotation_monaco_singler"
## [9] "sample_id_db" "_sample_name"Available assays
## # Source: SQL [?? x 2]
## # Database: DuckDB 0.8.1 [unknown@Linux 5.15.0-1041-azure:R 4.3.0/:memory:]
## assay n
## <chr> <dbl>
## 1 10x 3' v2 27
## 2 10x 3' v3 21
## 3 Visium Spatial Gene Expression 7
## 4 scRNA-seq 4
## 5 Seq-Well 2
## 6 sci-RNA-seq 1
## 7 Slide-seq 4
## 8 10x 5' v2 2
## 9 10x 5' v1 7
## 10 10x 3' v1 1
## # ℹ more rowsAvailable organisms
## # Source: SQL [1 x 2]
## # Database: DuckDB 0.8.1 [unknown@Linux 5.15.0-1041-azure:R 4.3.0/:memory:]
## organism n
## <chr> <dbl>
## 1 Homo sapiens 63Download single-cell RNA sequencing counts
The data can be provided as either “counts” or counts per million
“cpm” as given by the assays argument in the
get_single_cell_experiment() function. By default, the
SingleCellExperiment provided will contain only the
‘counts’ data.
Query raw counts
single_cell_counts <-
metadata |>
dplyr::filter(
ethnicity == "African" &
stringr::str_like(assay, "%10x%") &
tissue == "lung parenchyma" &
stringr::str_like(cell_type, "%CD4%")
) |>
get_single_cell_experiment()## ℹ Realising metadata.## ℹ Synchronising files## ℹ Downloading 0 files, totalling 0 GB## ℹ Reading files.## ℹ Compiling Single Cell Experiment.
single_cell_counts## class: SingleCellExperiment
## dim: 36229 1571
## metadata(0):
## assays(1): counts
## rownames(36229): A1BG A1BG-AS1 ... ZZEF1 ZZZ3
## rowData names(0):
## colnames(1571): ACACCAAAGCCACCTG_SC18_1 TCAGCTCCAGACAAGC_SC18_1 ...
## CAGCATAAGCTAACAA_F02607_1 AAGGAGCGTATAATGG_F02607_1
## colData names(56): sample_ cell_type ... updated_at_y original_cell_id
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):Query counts scaled per million
This is helpful if just few genes are of interest, as they can be compared across samples.
metadata |>
dplyr::filter(
ethnicity == "African" &
stringr::str_like(assay, "%10x%") &
tissue == "lung parenchyma" &
stringr::str_like(cell_type, "%CD4%")
) |>
get_single_cell_experiment(assays = "cpm")## ℹ Realising metadata.## ℹ Synchronising files## ℹ Downloading 0 files, totalling 0 GB## ℹ Reading files.## ℹ Compiling Single Cell Experiment.## class: SingleCellExperiment
## dim: 36229 1571
## metadata(0):
## assays(1): cpm
## rownames(36229): A1BG A1BG-AS1 ... ZZEF1 ZZZ3
## rowData names(0):
## colnames(1571): ACACCAAAGCCACCTG_SC18_1 TCAGCTCCAGACAAGC_SC18_1 ...
## CAGCATAAGCTAACAA_F02607_1 AAGGAGCGTATAATGG_F02607_1
## colData names(56): sample_ cell_type ... updated_at_y original_cell_id
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):Extract only a subset of genes
single_cell_counts <-
metadata |>
dplyr::filter(
ethnicity == "African" &
stringr::str_like(assay, "%10x%") &
tissue == "lung parenchyma" &
stringr::str_like(cell_type, "%CD4%")
) |>
get_single_cell_experiment(assays = "cpm", features = "PUM1")## ℹ Realising metadata.## ℹ Synchronising files## ℹ Downloading 0 files, totalling 0 GB## ℹ Reading files.## ℹ Compiling Single Cell Experiment.
single_cell_counts## class: SingleCellExperiment
## dim: 1 1571
## metadata(0):
## assays(1): cpm
## rownames(1): PUM1
## rowData names(0):
## colnames(1571): ACACCAAAGCCACCTG_SC18_1 TCAGCTCCAGACAAGC_SC18_1 ...
## CAGCATAAGCTAACAA_F02607_1 AAGGAGCGTATAATGG_F02607_1
## colData names(56): sample_ cell_type ... updated_at_y original_cell_id
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):Extracting counts as a Seurat object
If needed, the H5 SingleCellExperiment can be converted
into a Seurat object. Note that it may take a long time and use a lot of
memory depending on how many cells you are requesting.
single_cell_counts <-
metadata |>
dplyr::filter(
ethnicity == "African" &
stringr::str_like(assay, "%10x%") &
tissue == "lung parenchyma" &
stringr::str_like(cell_type, "%CD4%")
) |>
get_seurat()
single_cell_countsExercises
- Use
countandarrangeto get the number of cells per tissue in descending order.
# enter your code here- Use
dplyr-isms to group bytissueandcell_typeand get a tally of the highest number of cell types per tissue combination. What tissue has the most numerous type of cells?
# enter your code here- Spot some differences between the
tissueandtissue_harmonisedcolumns. Usecountto summarise.
# enter your code hereAnswer 3
## # Source: SQL [?? x 2]
## # Database: DuckDB 0.8.1 [unknown@Linux 5.15.0-1041-azure:R 4.3.0/:memory:]
## # Ordered by: -n
## tissue n
## <chr> <dbl>
## 1 cortex of kidney 36940
## 2 kidney 23549
## 3 lung parenchyma 16719
## 4 renal medulla 7729
## 5 respiratory airway 7153
## 6 blood 4248
## 7 bone marrow 4113
## 8 heart left ventricle 1454
## 9 transition zone of prostate 1140
## 10 lung 1137
## # ℹ more rows## # Source: SQL [?? x 2]
## # Database: DuckDB 0.8.1 [unknown@Linux 5.15.0-1041-azure:R 4.3.0/:memory:]
## # Ordered by: -n
## tissue_harmonised n
## <chr> <dbl>
## 1 kidney 68851
## 2 lung 25737
## 3 blood 4248
## 4 bone 4113
## 5 heart 1454
## 6 lymph node 1210
## 7 prostate 1156
## 8 intestine large 816
## 9 liver 793
## 10 thymus 753
## # ℹ more rowsTo see the full list of curated columns in the metadata, see the
Details section in the ?get_metadata documentation
page.
- Now that we are a little familiar with navigating the metadata,
let’s obtain a
SingleCellExperimentof 10X scRNA-seq counts ofcd8 temlungcells for females older than80withCOVID-19. Note: Use the harmonized columns, where possible.
# enter your code hereAnswer 4
metadata |>
dplyr::filter(
sex == "female" &
age_days > 80 * 365 &
stringr::str_like(assay, "%10x%") &
disease == "COVID-19" &
tissue_harmonised == "lung" &
cell_type_harmonised == "cd8 tem"
) |>
get_single_cell_experiment()## ℹ Realising metadata.## ℹ Synchronising files## ℹ Downloading 0 files, totalling 0 GB## ℹ Reading files.## ℹ Compiling Single Cell Experiment.## class: SingleCellExperiment
## dim: 36229 12
## metadata(0):
## assays(1): counts
## rownames(36229): A1BG A1BG-AS1 ... ZZEF1 ZZZ3
## rowData names(0):
## colnames(12): TCATCATCATAACCCA_1 TATCTGTCAGAACCGA_1 ...
## CCCTTAGCATGACTTG_1 CAGTTCCGTAGCGTAG_1
## colData names(56): sample_ cell_type ... updated_at_y original_cell_id
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):Session Info
## R version 4.3.0 (2023-04-21)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.2 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] dplyr_1.1.2 CuratedAtlasQueryR_0.99.6
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_1.8.7
## [3] magrittr_2.0.3 spatstat.utils_3.0-3
## [5] rmarkdown_2.23 zlibbioc_1.46.0
## [7] fs_1.6.2 ragg_1.2.5
## [9] vctrs_0.6.3 ROCR_1.0-11
## [11] memoise_2.0.1 spatstat.explore_3.2-1
## [13] RCurl_1.98-1.12 htmltools_0.5.5
## [15] S4Arrays_1.0.4 Rhdf5lib_1.22.0
## [17] rhdf5_2.44.0 sass_0.4.6
## [19] sctransform_0.3.5 parallelly_1.36.0
## [21] KernSmooth_2.23-22 bslib_0.5.0
## [23] htmlwidgets_1.6.2 desc_1.4.2
## [25] ica_1.0-3 plyr_1.8.8
## [27] plotly_4.10.2 zoo_1.8-12
## [29] cachem_1.0.8 igraph_1.5.0
## [31] mime_0.12 lifecycle_1.0.3
## [33] pkgconfig_2.0.3 Matrix_1.6-0
## [35] R6_2.5.1 fastmap_1.1.1
## [37] GenomeInfoDbData_1.2.10 MatrixGenerics_1.12.2
## [39] fitdistrplus_1.1-11 future_1.33.0
## [41] shiny_1.7.4.1 digest_0.6.33
## [43] colorspace_2.1-0 patchwork_1.1.2
## [45] S4Vectors_0.38.1 rprojroot_2.0.3
## [47] Seurat_4.3.0.1 tensor_1.5
## [49] irlba_2.3.5.1 GenomicRanges_1.52.0
## [51] textshaping_0.3.6 progressr_0.13.0
## [53] fansi_1.0.4 spatstat.sparse_3.0-2
## [55] polyclip_1.10-4 httr_1.4.6
## [57] abind_1.4-5 compiler_4.3.0
## [59] withr_2.5.0 DBI_1.1.3
## [61] highr_0.10 HDF5Array_1.28.1
## [63] duckdb_0.8.1 MASS_7.3-60
## [65] DelayedArray_0.26.6 tools_4.3.0
## [67] lmtest_0.9-40 httpuv_1.6.11
## [69] future.apply_1.11.0 goftest_1.2-3
## [71] glue_1.6.2 nlme_3.1-162
## [73] rhdf5filters_1.12.1 promises_1.2.0.1
## [75] grid_4.3.0 Rtsne_0.16
## [77] cluster_2.1.4 reshape2_1.4.4
## [79] generics_0.1.3 gtable_0.3.3
## [81] spatstat.data_3.0-1 tidyr_1.3.0
## [83] data.table_1.14.8 XVector_0.40.0
## [85] sp_2.0-0 utf8_1.2.3
## [87] BiocGenerics_0.46.0 spatstat.geom_3.2-2
## [89] RcppAnnoy_0.0.21 ggrepel_0.9.3
## [91] RANN_2.6.1 pillar_1.9.0
## [93] stringr_1.5.0 later_1.3.1
## [95] splines_4.3.0 lattice_0.21-8
## [97] deldir_1.0-9 survival_3.5-5
## [99] tidyselect_1.2.0 SingleCellExperiment_1.22.0
## [101] miniUI_0.1.1.1 pbapply_1.7-2
## [103] knitr_1.43 gridExtra_2.3
## [105] IRanges_2.34.1 SummarizedExperiment_1.30.2
## [107] scattermore_1.2 stats4_4.3.0
## [109] xfun_0.39 Biobase_2.60.0
## [111] matrixStats_1.0.0 stringi_1.7.12
## [113] lazyeval_0.2.2 yaml_2.3.7
## [115] evaluate_0.21 codetools_0.2-19
## [117] tibble_3.2.1 cli_3.6.1
## [119] uwot_0.1.16 xtable_1.8-4
## [121] reticulate_1.30 systemfonts_1.0.4
## [123] munsell_0.5.0 jquerylib_0.1.4
## [125] GenomeInfoDb_1.36.1 Rcpp_1.0.11
## [127] spatstat.random_3.1-5 globals_0.16.2
## [129] dbplyr_2.3.3 png_0.1-8
## [131] parallel_4.3.0 ellipsis_0.3.2
## [133] blob_1.2.4 assertthat_0.2.1
## [135] pkgdown_2.0.7 ggplot2_3.4.2
## [137] bitops_1.0-7 listenv_0.9.0
## [139] viridisLite_0.4.2 scales_1.2.1
## [141] ggridges_0.5.4 SeuratObject_4.1.3
## [143] leiden_0.4.3 purrr_1.0.1
## [145] crayon_1.5.2 rlang_1.1.1
## [147] cowplot_1.1.1Acknowledgements
Thank you to Stefano Mangiola and his team for developing CuratedAtlasQueryR and graciously providing the content from their vignette. Make sure to keep an eye out for their publication for proper citation. Their bioRxiv paper can be found at https://www.biorxiv.org/content/10.1101/2023.06.08.542671v1.