Installing and Managing _Bioconductor_ Packages
Marcel Ramos
Roswell Park Comprehensive Cancer Center, Buffalo, NYMartin Morgan
Roswell Park Comprehensive Cancer Center, Buffalo, NYSource:
vignettes/BiocManager.Rmd
BiocManager.RmdIntroduction
Use the BiocManager package to install and manage packages from the Bioconductor project for the statistical analysis and comprehension of high-throughput genomic data.
Current Bioconductor packages are available on a ‘release’ version intended for every-day use, and a ‘devel’ version where new features are introduced. A new release version is created every six months. Using the BiocManager package helps users install packages from the same release.
Basic use
Installing R
We recommend using the current ‘release’ version of R. Follow instructions for installing R.
Installing BiocManager
Use standard R installation procedures to install the BiocManager package. This command is required only once per R installation.
install.packages("BiocManager", repos = "https://cloud.r-project.org")Installing Bioconductor, CRAN, or GitHub packages
Install Bioconductor (or CRAN) packages with
Installed packages can be updated to their current version with
BiocManager::install()Previous releases
To install CRAN package versions consistent with previous releases of Bioconductor, use the BiocArchive package. BiocArchive enables contemporary installations of CRAN packages with out-of-date Bioconductor releases using Posit Public Package Manager.
Version and validity of installations
Use version() to discover the version of
Bioconductor currently in use.
BiocManager::version()Bioconductor packages work best when they are all from the
same release. Use valid() to identify packages that are
out-of-date or from unexpected versions.
BiocManager::valid()valid() returns an object that can be queried for
detailed information about invalid packages, as illustrated in the
following screen capture
> v <- valid()
Warning message:
6 packages out-of-date; 0 packages too new
> names(v)
[1] "out_of_date" "too_new"
> head(v$out_of_date, 2)
Package LibPath
bit "bit" "/home/mtmorgan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
ff "ff" "/home/mtmorgan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
Installed Built ReposVer Repository
bit "1.1-12" "3.5.0" "1.1-13" "https://cloud.r-project.org/src/contrib"
ff "2.2-13" "3.5.0" "2.2-14" "https://cloud.r-project.org/src/contrib"
>Available packages
Packages available for your version of Bioconductor can be
discovered with available(); the first argument can be used
to filter package names based on a regular expression, e.g., ‘BSgenome’
package available for Homo sapiens
avail <- BiocManager::available()
length(avail) # all CRAN & Bioconductor packages
BiocManager::available("BSgenome.Hsapiens") # BSgenome.Hsapiens.* packagesQuestions about installing and managing Bioconductor packages should be addressed to the Bioconductor support site.
Updating old packages
The user can often get an update packages prompt similar to:
> BiocManager::install("AnVIL")
Bioconductor version 3.18 (BiocManager 1.30.22), R 4.3.1 (2023-06-16)
Old packages: 'AnnotationDbi', 'arrow', 'basilisk.utils', 'beachmat',
'BiocParallel', 'BiocSingular', 'biocthis', 'Biostrings', 'bluster',
'DelayedArray', 'downlit', 'edgeR', 'GenomeInfoDb', 'GenomicFeatures',
'IRanges', 'limma', 'pkgbuild', 'pkgload', 'processx', 'RcppSpdlog', 'rhdf5',
'rstudioapi', 'SingleR', 'testthat', 'usethis', 'xml2', 'KernSmooth',
'Matrix', 'mgcv'
Update all/some/none? [a/s/n]:A decision should be made regarding updating packages. In the following section we will detail the rationale, pros, and cons of updating packages.
(An initial draft of this section was produced by ChatGPT on 28 August 2023)
Rationale
Package updates often include bug fixes, improvements in functionality, and optimizations. By updating, the user can ensure that they are benefiting from the latest enhancements and fixes in the packages they use.
Pros of updating
- Bug Fixes: Updating packages can resolve known issues and bugs present in older versions. This can lead to more accurate and reliable analyses.
- Performance: Package updates might come with performance optimizations that can lead to faster execution of code and analyses.
- New Features: Updated packages might introduce new features, functions, and capabilities that can enhance your workflow and analysis options.
- Compatibility: Newer packages are often developed with compatibility in mind. Using outdated packages might lead to compatibility issues with other packages or R itself.
- Security: In some cases, package updates address security vulnerabilities. Keeping packages up to date can help maintain the security of the computational environment.
- Documentation: Newer versions of packages might come with updated documentation that reflects recent changes and improvements in package functionality.
Cons of updating
- Code Breakage: Updates can introduce changes in package behavior, function signatures, or syntax. This can potentially break existing code if it relies on the previous behavior.
- Version Conflicts: Updates to one package might trigger dependencies on updated versions of other packages. This can lead to conflicts if those updated versions are not compatible with other parts of your workflow.
- Workflow Disruption: When you update packages, you might need to retest and validate your analyses to ensure that the changes in package behavior don’t affect your results.
- Learning Curve: New features and changes introduced in updated packages might require you to invest time in understanding how to use them effectively.
- Temporary Instability: Right after a major update, the new version might not be as stable as the previous one, leading to unexpected behavior.
Balancing the Decision
- Consider how critical the package is to your analysis. If a package is central to your workflow, updating might be more important.
- Read the release notes of the package to understand what changes are introduced in the update.
- Consider using a separate environment (e.g., Docker container) for testing updates before applying them to the main analysis environment.
- Make sure to backup code and data before performing updates.
- Check if there are any compatibility issues with other packages you are using.
Archived CRAN packages
In the event that a package is no longer available on CRAN, it may be desirable to install an archived package, especially if it is assumed that the package will return to an unarchived state at a later date. Users who wish to protect their systems from the consequences of these state changes can install archived packages by using the CRANhaven repository. This repository contains archived versions of CRAN packages for up to five weeks or until they are unarchived on CRAN. To enable archived CRAN package installations, the user can run the following command:
Advanced use
Changing version
Use the version= argument to update all packages to a
specific Bioconductor version
BiocManager::install(version="3.7")Bioconductor versions are associated with specific R versions, as summarized here. Attempting to install a version of Bioconductor that is not supported by the version of R in use leads to an error; using the most recent version of Bioconductor may require installing a new version of R.
> BiocManager::install(version="3.9")
Error: Bioconductor version '3.9' requires R version '3.6'; see
https://bioconductor.org/installA special version, version="devel", allows use of
Bioconductor packages that are under development.
Unsupported R / Bioconductor versions
The main purpose of BiocManager is to ensure that users install the
version of Bioconductor appropriate for their version of
R. Use the environment variable R_BIOC_VERSION to
install any version of Bioconductor on any version of
R. Thus R version 4.3.0 and Bioconductor
version 3.19 are not compatible…
> BiocManager::install(version = "3.19")
Error: Bioconductor version '3.19' requires R version '4.4'; use
`version = '3.18'` with R version 4.3; see
https://bioconductor.org/install…but the version can be forced with
> Sys.setenv(R_BIOC_VERSION="3.19")
> BiocManager::install(version = "3.19")
...
Bioconductor version 3.19 (BiocManager 1.30.23), R 4.3.0 (2023-04-21)Note. Compatibility of Bioconductor with a mismatched version of R is not guaranteed and support is not provided for such installations.
Managing multiple versions
It is possible to have multiple versions of Bioconductor installed on the same computer. A best practice is to create an initial R installation. Then create and use a library for each version of Bioconductor. The library will contain all Bioconductor, CRAN, and other packages for that version of Bioconductor. We illustrate the process assuming use of Bioconductor version 3.7, available using R version 3.5
Create a directory to contain the library (replace
USER_NAME with your user name on Windows)
- Linux:
~/R/3.5-Bioc-3.7 - macOS:
~/Library/R/3.5-Bioc-3.7/library - Windows:
C:\Users\USER_NAME\Documents\R\3.5-Bioc-3.7
Set the environment variable R_LIBS_USER to this
directory, and invoke R. Command line examples for Linux
are
- Linux:
R_LIBS_USER=~/R/3.5-Bioc-3.7 R - macOS:
R_LIBS_USER=~/Library/R/3.5-Bioc-3.7/library R - Windows:
cmd /C "set R_LIBS_USER=C:\Users\USER_NAME\Documents\R\3.5-Bioc-3.7 && R"
Once in R, confirm that the version-specific library path has been set
On Linux and macOS, create a bash alias to save typing, e.g.,
- Linux:
alias Bioc3.7='R_LIBS_USER=~/R/3.5-Bioc-3.7 R' - macOS:
alias Bioc3.7='R_LIBS_USER=~/Library/R/3.5-Bioc-3.7/library R'
Invoke these from the command line as Bioc3.7.
On Windows, create a shortcut. Go to My Computer and navigate to a directory that is in your PATH. Then right-click and choose New->Shortcut. In the “type the location of the item” box, put:
cmd /C "set R_LIBS_USER=C:\Users\USER_NAME\Documents\R\3.5-Bioc-3.7 && R"Click “Next”. In the “Type a name for this shortcut” box, type
Bioc-3.7.
Using the same R version
Note that managing multiple versions of R and Bioconductor is important even when the Bioconductor versions use the same R version. The Bioconductor libraries should be kept in separate directories to avoid conflicts between packages. For example, Bioconductor versions 3.7 and 3.8 both use R version 3.5. To manage packages from these versions, create two separate library directories. Using the same setup as shown above, the second library directory would be:
- Linux:
~/R/3.5-Bioc-3.8 - macOS:
~/Library/R/3.5-Bioc-3.8/library - Windows:
C:\Users\USER_NAME\Documents\R\3.5-Bioc-3.8
It is also convenient to set a separate alias or shortcut for this version:
- Linux:
alias Bioc3.8='R_LIBS_USER=~/R/3.5-Bioc-3.8 R' - macOS:
alias Bioc3.8='R_LIBS_USER=~/Library/R/3.5-Bioc-3.8/library R'
On Windows, create a shortcut. Go to My Computer and navigate to a directory that is in your PATH. Then right-click and choose New->Shortcut. In the “type the location of the item” box, put:
- Windows:
cmd /C "set R_LIBS_USER=C:\Users\USER_NAME\Documents\R\3.5-Bioc-3.8 && R"
Now there are two sets of aliases or shortcuts, one for each version
of Bioconductor. This strategy can be extended to any number of versions
of Bioconductor as long as the corresponding R version is installed on
the system. When working with separate R versions, the path to the
R executable must be specified in the alias or
shortcut.
Keep in mind that mixing packages from different versions of Bioconductor can lead to unexpected behavior and errors.
Offline use
Offline use of BiocManager is possible for organizations and users that would like to provide access to internal repositories of Bioconductor packages while enforcing appropriate version checks between Bioconductor and R. For offline use, organizations and users require the following steps:
-
Use
rsyncto create local repositories of CRAN and Bioconductor. Tell R about these repositories using (e.g., in a site-wide.Rprofile, see?.Rprofile).options( repos = c(CRAN_mirror = "file:///path/to/CRAN-mirror"), BioC_mirror = "file:///path/to/Bioc-mirror" )Validate repository setting by reviewing the output of
repositories(). -
Create an environment variable or option, e.g.,
options( BIOCONDUCTOR_ONLINE_VERSION_DIAGNOSIS = FALSE ) -
Use
install.packages()to bootstrap the BiocManager installation.install.package(c("BiocManager", "BiocVersion"))
BiocManager can then be used for subsequent installations, e.g.,
BiocManager::install(c("ggplot2", "GenomicRanges")).
Offline config.yaml
BiocManager also expects to reference an online
configuration yaml file for Bioconductor version validation at
https://bioconductor.org/config.yaml. With offline use,
users are expected to either host this file locally or provide their
config.yaml version. The package allows either an
environment variable or R-specific option to locate this file, e.g.,
``` r
options(
BIOCONDUCTOR_CONFIG_FILE = "file:///path/to/config.yaml"
)
```How it works
BiocManager’s job is to make sure that all packages are installed from the same Bioconductor version, using compatible R and CRAN packages. However, R has an annual release cycle, whereas Bioconductor has a twice-yearly release cycle. Also, Bioconductor has a ‘devel’ branch where new packages and features are introduced, and a ‘release’ branch where bug fixes and relative stability are important; CRAN packages do not distinguish between devel and release branches.
In the past, one would install a Bioconductor package by
evaluating the command source("https://.../biocLite.R") to
read a file from the web. The file contained an installation script that
was smart enough to figure out what version of R and
Bioconductor were in use or appropriate for the person invoking
the script. Sourcing an executable script from the web is an obvious
security problem. In 2024, the script was removed and is no longer in
use.
Our solution is to use a CRAN package BiocManager, so that users install from pre-configured CRAN mirrors rather than typing in a URL and sourcing from the web.
But how does a CRAN package know what version of Bioconductor is in use? Can we use BiocManager? No, because we don’t have enough control over the version of BiocManager available on CRAN, e.g., everyone using the same version of R would get the same version of BiocManager and hence of Bioconductor. But there are two Bioconductor versions per R version, so that does not work!
BiocManager could write information to a cache on the user disk, but this is not a robust solution for a number of reasons. Is there any other way that R could keep track of version information? Yes, by installing a Bioconductor package (BiocVersion) whose sole purpose is to indicate the version of Bioconductor in use.
By default, BiocManager installs the BiocVersion package
corresponding to the most recent released version of
Bioconductor for the version of R in use. At the time
this section was written, the most recent version of R is R-3.6.1,
associated with Bioconductor release version 3.9. Hence on
first use of BiocManager::install() we see BiocVersion
version 3.9.0 being installed.
> BiocManager::install()
Bioconductor version 3.9 (BiocManager 1.30.4), R 3.6.1 Patched (2019-07-06
r76792)
Installing package(s) 'BiocVersion'
trying URL 'https://bioconductor.org/packages/3.9/bioc/src/contrib/\
BiocVersion_3.9.0.tar.gz'
...Requesting a specific version of Bioconductor updates, if possible, the BiocVersion package.
> ## 3.10 is available for R-3.6
> BiocManager::install(version="3.10")
Upgrade 3 packages to Bioconductor version '3.10'? [y/n]: y
Bioconductor version 3.10 (BiocManager 1.30.4), R 3.6.1 Patched (2019-07-06
r76792)
Installing package(s) 'BiocVersion'
trying URL 'https://bioconductor.org/packages/3.10/bioc/src/contrib/\
BiocVersion_3.10.1.tar.gz'
...
> ## back down again...
> BiocManager::install(version="3.9")
Downgrade 3 packages to Bioconductor version '3.9'? [y/n]: y
Bioconductor version 3.9 (BiocManager 1.30.4), R 3.6.1 Patched (2019-07-06
r76792)
Installing package(s) 'BiocVersion'
trying URL 'https://bioconductor.org/packages/3.9/bioc/src/contrib/\
BiocVersion_3.9.0.tar.gz'
...Answering n to the prompt to up- or downgrade packages
leaves the installation unchanged, since this would immediately create
an inconsistent installation.
Troubleshooting
Package not available
(An initial draft of this section was produced by ChatGPT on 25 May 2023)
A user failed to install the ‘celldex’ package on 25 May 2023. A transcript of the R session is as follows:
> BiocManager::version()
[1] '3.18'
> BiocManager::install("celldex")
Bioconductor version 3.18 (BiocManager 1.30.20), R 4.3.0 Patched (2023-05-01
r84362)
Installing package(s) 'celldex'
Warning message:
package 'celldex' is not available for Bioconductor version '3.18'
A version of this package for your version of R might be available elsewhere,
see the ideas at
https://cran.r-project.org/doc/manuals/r-patched/R-admin.html#Installing-packagesThe availability of specific packages within Bioconductor can depend on various factors, including simple errors in entering the package name, the package’s development status, maintenance, and compatibility with the latest version of Bioconductor, as well as the availability of CRAN packages that the Bioconductor package depends on.
Package Name: R package names are case sensitive and must be
spelt correctly, so using BiocManager::install("Celldex")
(with a capital C) or
BiocManager::install("celdex") (with only one
l) would both fail to install celldex;
R will sometimes suggest the correct name.
CRAN Packages: BiocManager::install() tries to
install packages from CRAN and from Bioconductor. Check that
the package is not a CRAN package by trying to visit the CRAN ‘landing
page’
https://cran.R-project.org/package=celldex
If this page is found, then the package is a CRAN package; see the R-admin manual section on troubleshooting CRAN package installations.
Check also that the package is not a CRAN package that has been ‘archived’ and no longer available by trying to visit
https://cran.R-project.org/src/contrib/Archive/celldex/
If this page exists but the ‘landing page’ does not, this means that the package has been removed from CRAN. While it is possible to install archived packages, usually the best course of action is to identify alternative packages to accomplish the task you are interested in. This is especially true if the ‘Last modified’ date of the most recent archived package is more than several months ago.
Compatibility: A Bioconductor package must be available for the specific version of Bioconductor you are using. Try visiting the ‘landing page’ of the package for your version of Bioconductor, e.g., for Bioconductor version 3.18 and package celldex
If this landing page does not exist, then the package is not available for your version of Bioconductor.
Users may sometimes have an out-of-date version of R or Bioconductor installed; this may be intentional (e.g., to ensure reproducibility of existing analyses) or simply because Bioconductor has not yet been updated. Try visiting the current release landing page
If the release landing page exists, and it is not important that you continue using the out-of-date version of Bioconductor, consider updating R (if necessary) and Bioconductor to the current release versions using instructions at the top of this document.
Packages recently contributed to Bioconductor are added to the ‘devel’ branch, whereas most users are configured to use the ‘release’ branch. Try visiting the ‘devel’ landing page
If only the devel landing page exists, then consider updating your installation to use the development version of Bioconductor. Note that the development version is not as stable as the release version, so should not be used for time-critical or ‘production’ analysis.
It may be that the package you are interested in has been removed from Bioconductor. Check this by visiting
If the package has been removed, the best course of action is to identify alternative packages to accomplish the task you are interested in.
Maintenance and Operating System Availability: A package may be included in the release or devel version of Bioconductor, but currently unavailable because it requires maintenance. This might be indicated by a red ‘build’ badge as in the image below (details of the build problem are available by clicking on the badge). The build error usually requires that the package maintainer correct an issue with their package; the maintainer and email address are listed on the package landing page.
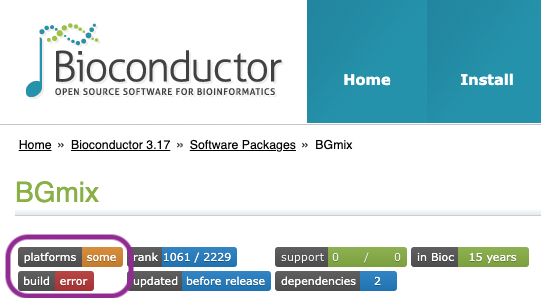
A small number of Bioconductor packages are not available on all operating systems. An orange ‘platforms’ badge indicates this. Click on the badge to be taken to the ‘Package Archives’ section of the landing page; BGmix is not supported on Windows, and not available on ‘Intel’ macOS because of build-related errors. Consider using an alternative operating system if the package is essential to your work

Packages with landing pages from older releases but not available for your operating system cannot be updated by the maintainer. If the package is available in the current release and for your operating system, consider updating to the current release of Bioconductor.
Cannot load BiocManager
After updating R (e.g., from R version 3.5.x to
R version 3.6.x at the time of writing this) and trying to load
BiocManager, R replies
Error: .onLoad failed in loadNamespace() for 'BiocManager', details:
call: NULL
error: Bioconductor version '3.8' requires R version '3.5'; see
https://bioconductor.org/installThis problem arises because BiocManager uses a second
package, BiocVersion, to indicate the version of
Bioconductor in use. In the original installation,
BiocManager had installed BiocVersion
appropriate for R version 3.5. With the update, the version of
Bioconductor indicated by BiocVersion is no longer
valid – you’ll need to update BiocVersion and all
Bioconductor packages to the most recent version available for
your new version of R.
The recommended practice is to maintain a separate library for each R and Bioconductor version. So instead of installing packages into R’s system library (e.g., as ‘administrator’), install only base R into the system location. Then use aliases or other approaches to create R / Bioconductor version-specific installations. This is described in the section on maintaining multiple versions of R and Bioconductor.
Alternatively, one could update all Bioconductor packages in
the previous installation directory. The problem with this is that the
previous version of Bioconductor is removed, compromising the
ability to reproduce earlier results. Update all Bioconductor
packages in the previous installation directory by removing all
versions of BiocVersion
remove.packages("BiocVersion") # repeat until all instances removedThen install the updated BiocVersion, and update all
Bioconductor packages; answer ‘yes’ when you are asked to
update a potentially large number of Bioconductor packages.
BiocManager::install()Confirm that the updated Bioconductor is valid for your version of R
BiocManager::valid()Timeout during package download
Large packages can take a long time to downloaded over poor internet
connects. The BiocManager package sets the time limit to 300 seconds,
using options(timeout = 300). Only part of a package may
download, e.g., only 15.1 of 79.4 MB in the example below
trying URL 'https://bioconductor.org/packages/3.12/data/annotation/src/contrib/org.Hs.eg.db_3.12.0.tar.gz'
Content type 'application/x-gzip' length 83225518 bytes (79.4 MB)
=========
downloaded 15.1 MBor perhaps with a warning (often difficult to see in the output)
Error in download.file(url, destfile, method, mode = "wb", ...) :
...
...: Timeout of 300 seconds was reached
...Try increasing the download timeout, e.g,
options(timeout = 600).
Multiple BiocVersion installations
One potential problem occurs when there are two or more
.libPaths(), with more than one BiocVersion package
installed. This might occur for instance if a ‘system administrator’
installed BiocVersion, and then a user installed their own version. In
this circumstance, it seems appropriate to standardize the installation
by repeatedly calling remove.packages("BiocVersion") until
all versions are removed, and then installing the desired version.
Errors determining Bioconductor version
An essential task for BiocManager is to determine that the version of Bioconductor is appropriate for the version of R. Several errors can occur when this task fails.
Bioconductor version cannot be determined; no internet connection? When the Bioconductor version cannot be obtained from the version map hosted at https://bioconductor.org/config.yaml, this error will occur. It may be a result of poor internet connectivity or offline use. See the offline config.yaml section above.
Bioconductor version cannot be validated; no internet connection? Usually occurs when the map is unable to be downloaded possibly due to a missing
BIOCONDUCTOR_CONFIG_FILE. For offline use, a copy of the configuration file should be downloaded and its address set to the environment variable or option.Bioconductor version map cannot be validated; is it misconfigured? On rare occasion, the version map hosted at https://bioconductor.org/config.yaml may be misconfigured. The check ensures that all the version name tags, i.e., out-of-date, release, devel, and future are in the map.
Bioconductor version cannot be validated; is type input mis-specified? The type input refers to the version name inputs, mainly release and devel. This error is chiefly due to internal logic and is not due to user error. Please open a GitHub issue.
Session information
## R version 4.5.2 (2025-10-31)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] BiocStyle_2.38.0
##
## loaded via a namespace (and not attached):
## [1] digest_0.6.38 desc_1.4.3 R6_2.6.1
## [4] bookdown_0.45 fastmap_1.2.0 xfun_0.54
## [7] cachem_1.1.0 knitr_1.50 htmltools_0.5.8.1
## [10] rmarkdown_2.30 lifecycle_1.0.4 cli_3.6.5
## [13] sass_0.4.10 pkgdown_2.2.0 textshaping_1.0.4
## [16] jquerylib_0.1.4 systemfonts_1.3.1 compiler_4.5.2
## [19] tools_4.5.2 ragg_1.5.0 evaluate_1.0.5
## [22] bslib_0.9.0 yaml_2.3.10 BiocManager_1.30.27
## [25] jsonlite_2.0.0 rlang_1.1.6 fs_1.6.6